Localized chemical bonding may be defined as bonding in which the electrons are shared by two and only two nuclei.
Wave mechanics is predicated on the foundational axiom that electrons exhibit wavelike behavior, evidenced by phenomena such as diffraction, thereby necessitating the formulation of a wave equation applicable to them. This conceptual framework parallels the theoretical treatment of other waveforms, such as electromagnetic and acoustic waves, which are also governed by corresponding wave equations. The mathematical formalism that encapsulates the behavior of electrons is encapsulated in the Schrödinger equation. For a univalent electron system, this equation assumes the following form:
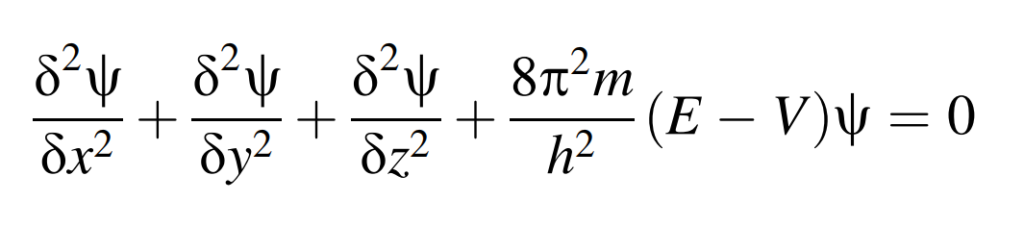
Within this equation, m denotes the electron’s mass, E represents its total energy, V signifies its potential energy, and h is Planck’s constant. Physically, the wave function delineates the probabilistic amplitude, with its square yielding the probability density of locating the electron at any spatial coordinates x, y, and z, where the origin is positioned at the nucleus. For poly-electronic systems, the equation retains its foundational structure but becomes exponentially more intricate in its complexity.
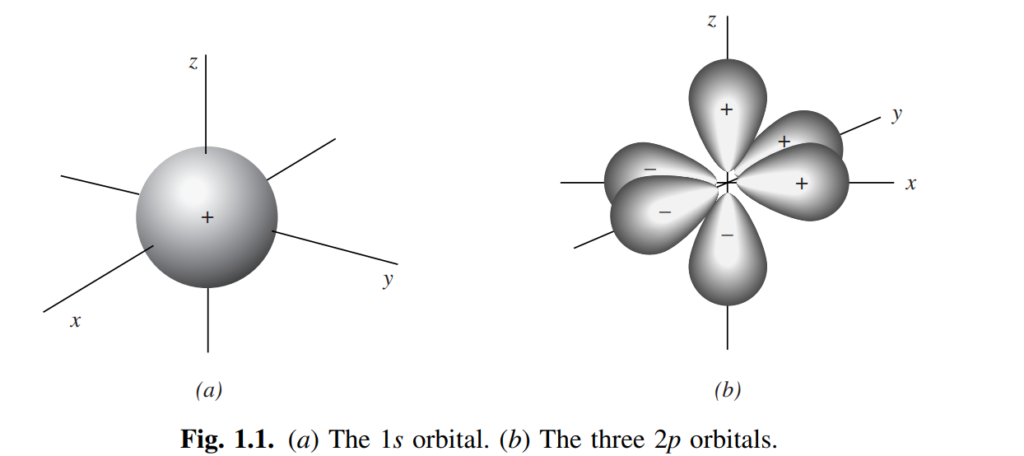
The Schrödinger equation is a differential equation, signifying that its solutions are themselves equations, albeit not differential in nature. These solutions are relatively straightforward equations that can be graphically represented. Such graphical representations, three-dimensional depictions illustrating electron density, are termed orbitals or electron clouds. Many students are acquainted with the configurations of s and p atomic orbitals (refer to Fig. 1.1). Notably, each p orbital exhibits a node—a spatial region where the probability of locating the electron is minuscule.(Wave-mechanical calculations using the Schrödinger equation indicate that the probability of finding an electron in a node is zero. However, when relativistic effects are considered, Dirac has shown that nodes do have a very small electron density (Powell, R.E., J. Chem. Educ., 1968, 45, 558; Ellison, F.O. and Hollingsworth, C.A., J. Chem. Educ., 1976, 53, 767; McKelvey, D.R., J. Chem. Educ., 1983, 60, 112; Nelson, P.G., J. Chem. Educ., 1990, 67, 643). For a general review on relativistic effects on chemical structures, see Pyykko, P., Chem. Rev., 1988, 88, 563.For simple systems like the H2 molecule or the He atom, there are approximate solutions that are nearly as accurate as exact solutions (Roothaan, C.C.J.; Weiss, A.W., Rev. Mod. Phys., 1960, 32, 194; Kolos, W.; Roothaan, C.C.J., Rev. Mod. Phys., 1960, 32, 219). For a review, see Clark, R.G.; Stewart, E.T., Q. Rev. Chem. Soc., 1970, 24, 95.)
Additionally, in Fig. 1.1, some orbital lobes are marked with positive and negative signs. These signs do not indicate electrical charges, as both lobes of an electron cloud inherently carry negative charge. Instead, they denote the signs of the wave function (ψ). When an orbital is bisected by a node, ψ consistently exhibits opposite signs on either side of the node.
According to the Pauli exclusion principle, no more than two electrons can occupy any given orbital, and they must possess opposite spins. Unfortunately, the Schrödinger equation admits exact solutions only for single-electron systems, such as the hydrogen atom. If exact solutions were attainable for multi-electron molecules, we would possess an exact depiction of the orbitals’ shapes (particularly for the crucial ground state) and the energy associated with each orbital. Given that exact solutions are elusive, substantial approximations are necessitated.
There are two primary methods of approximation:
- The Molecular-orbital method and;
- Valence-bond method
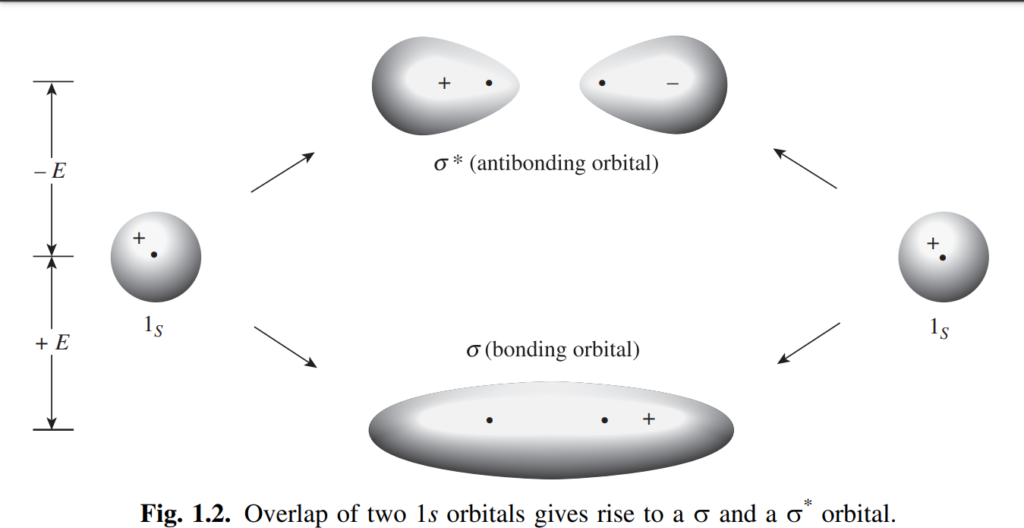
In the molecular-orbital method, bonding is perceived to emerge from the overlap of atomic orbitals. When multiple atomic orbitals overlap, they amalgamate to form an equivalent number of new orbitals, referred to as molecular orbitals. Molecular orbitals differ fundamentally from atomic orbitals in that they encompass the nuclei of two or more atoms, rather than being confined to a single atom. In localized bonding, the number of overlapping atomic orbitals is typically two (each containing one electron), resulting in the formation of two molecular orbitals. One of these, termed a bonding orbital, possesses lower energy than the original atomic orbitals (a prerequisite for bond formation), while the other, known as an antibonding orbital, has higher energy. Orbitals with lower energy levels are populated first. Given that the two initial atomic orbitals each contained one electron, both electrons can now occupy the new molecular bonding orbital, as any orbital can accommodate two electrons. The antibonding orbital remains vacant in the ground state. The extent of overlap directly correlates with bond strength, although complete overlap is hindered by nuclear repulsion.
Figure 1.2 illustrates the bonding and antibonding orbitals that emerge from the overlap of two 1s electrons. Notably, the antibonding orbital features a node between the nuclei, resulting in minimal electron density in that region, thereby diminishing its bonding efficacy. Molecular orbitals formed by the overlap of two atomic orbitals with their electron density centers aligned along the axis common to the two nuclei are termed sigma (σ) orbitals, and the resultant bonds are known as sigma bonds. Corresponding antibonding orbitals are denoted as σ*. Sigma orbitals can form not only through the overlap of two s orbitals but also via the overlap of any types of atomic orbitals (s, p, d, or f), whether identical or disparate, provided that the overlapping lobes have the same sign: a positive s orbital can bond only with another positive s orbital or with a positive lobe of a p, d, or f orbital. Regardless of the types of atomic orbitals they originate from, sigma orbitals can generally be approximated as ellipsoidal in shape.
Orbitals are frequently identified by their symmetry properties. The s orbital of hydrogen is often denoted as σg, where the ‘g’ signifies ‘gerade.’ A gerade orbital maintains its sign when inverted through its center of symmetry. Conversely, the σ* orbital is ‘ungerade’ (designated as σu), indicating that it changes sign when inverted through its center of symmetry.